Absorption versus Adsorption: High-Throughput Computation of Impurities in 2D Materials
Joel Davidsson, Fabian Bertoldo, Kristian S. Thygesen, Rickard Armiento
arXiv:2207.05353 (cond-mat)
If you are using data from this database in your research, please cite the following paper:
Absorption versus Adsorption: High-Throughput Computation of Impurities in 2D Materials
Joel Davidsson, Fabian Bertoldo, Kristian S. Thygesen, Rickard Armiento
arXiv:2207.05353 (cond-mat)
Please also cite the related DOI for the actual data:
Interstitial and Adsorbate Structure Database
Joel Davidsson, Fabian Bertoldo, Kristian S. Thygesen, Rickard Armiento
DOI 10.11583/DTU.19692238
Download database: imp2d.db
This database contains structural, thermodynamic, and electronic data of more than 17,500 single impurity defects over a set of 53 experimentally synthesized 2D monolayers. The defect structures (interstitials, adsorption sites) were set up with the ASE DefectBuilder module. The computational workflow was implemented with the httk High-Throughput Toolkit. 12,813 of the overall 17,364 systems have reached full ionic convergence as described in the publication above.
If a parameter is not specified at a given step, its value equals that of the last step where it was specified:
Workflow step(s) |
Parameters |
|---|---|
Initial relaxation |
Electronic structure code: VASP; xc functional = PBE; \(k\)-point grid: Gamma-point only; spin-polarized; PW energy cutoff = 600 eV; kinetic energy cutoff = 900 eV; electronic tolerance (energy) = 10`^{-4}`; ionic tolerance (energy) = 0.005 eV; FFT grid = 3/2 |
Final relaxation |
Electronic structure code: VASP; xc functional = PBE; \(k\)-point grid: Gamma-point only; spin-polarized; PW energy cutoff = 600 eV; kinetic energy cutoff = 900 eV; electronic tolerance (energy) = 10`^{-6}`; ionic tolerance (energy) = 0.0001 eV; FFT grid = 2 |
The project has received funding from the Danish National Research Foundation’s Center for Nanostructured Graphene (CNG) and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program Grant No. 773122 (LIMA) and Grant agreement No. 951786 (NOMAD CoE). We acknowlege funding from the Swedish eScience Centre (SeRC) and support from the Swedish REsearch Council (VR) Grant No. 2020-05402. The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC and PDC partially funded by the Swedish Research Council through Grant Agreement No. 2018-05973.
Version |
rows |
comment |
2022-07-12 |
17364 |
Initial release |
key |
description |
unit |
|---|---|---|
|
Host chemical formula |
|
|
Impurity species |
|
|
Impurity type |
|
|
Formation energy |
eV |
|
Defect setup position |
|
|
Depth parameter |
|
|
Extension factor |
|
|
Total energy |
eV |
|
Total energy convergence |
eV |
|
Magnetic moment |
|
|
Calculation supercell |
|
|
Host spacegroup symbol |
|
|
Relaxation converged? |
|
|
System name |
|
|
Total energy, first WF step |
eV |
|
Total energy convergence, first WF step |
eV |
|
Dopant chemical potential |
eV |
|
Host energy |
eV |
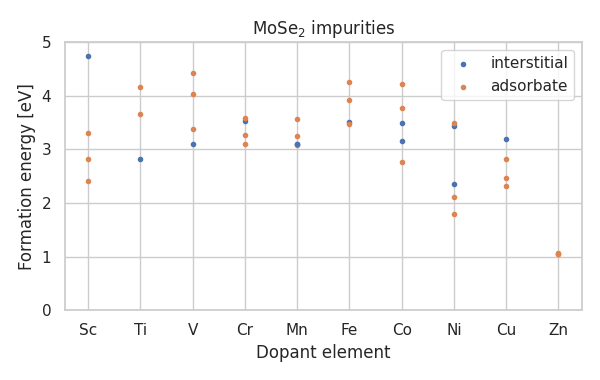
The following Python script shows how to plot the formation energies of period 4 transition metal impurities (for different interstitial and adsorption sites).
# creates: formation_energies_MoSe2.png
from ase.data import atomic_numbers
from ase.db import connect
import matplotlib.pyplot as plt
def get_plot_data(db, hosts, dopants):
"""Return data needed for subsequent plotting."""
eforms = []
numbers = []
defecttypes = []
# loop over all given host materials
for host in hosts:
# loop over all given dopants
for dopant in dopants:
# only include data that is fully converged and relaxed
for row in db.select(host=host, dopant=dopant, converged=True):
# convert dopant string to atomic number and append to list
numbers.append(atomic_numbers[dopant])
# extract formation energy and defecttype
# (interstitial or adsorbate)
eforms.append(row.eform)
defecttypes.append(row.defecttype)
return eforms, numbers, defecttypes
def plot(filename):
# connect to the 'Impurities in 2D Materials database'
db = connect('imp2d.db')
# extract plotting data for group 4 transition metal doped MoSe2
hosts = ['MoSe2']
dopants = ['Sc', 'Ti', 'V', 'Cr', 'Mn', 'Fe', 'Co', 'Ni', 'Cu', 'Zn']
eforms, numbers, defecttypes = get_plot_data(db, hosts, dopants)
# the actual plotting is happening here:
# matplotlib setup
fig, ax = plt.subplots(figsize=(6, 3.8))
# plot the formation energies for the period 4 TM-impurities in MoS2
for i in range(len(eforms)):
if defecttypes[i] == 'interstitial':
color = 'C0'
elif defecttypes[i] == 'adsorbate':
color = 'C1'
# plot formation energy versus dopant element
ax.scatter(numbers[i], eforms[i], marker='.', color=color)
# set title
ax.set_title(r'MoSe$_2$ impurities')
# set up the legend
ax.scatter([], [], marker='.', color='C0',
label='interstitial')
ax.scatter([], [], marker='.', color='C1',
label='adsorbate')
ax.legend()
# fix xticks
ax.set_xticks(list(set(numbers)))
ax.set_xticklabels(dopants)
# set x- and y-label
ax.set_xlabel(r'Dopant element')
ax.set_ylabel(r'Formation energy [eV]')
# set plot limits
ax.set_ylim(0, 5)
# save figure
plt.tight_layout()
plt.savefig(filename)
# plt.show()
plot('formation_energies_MoSe2.png')
This produces the figure
The defect setup was inspired by the work of J. Davidsson for ADAQ and generalized in the ASE DefectBuilder. Find more information, explanations, and some code examples in the documentation of the DefectBuilder.
The workflow was executed using httk.