CMR¶
Each CMR project in the table below consists of an ASE-database and a project page describing the data and showing examples of how to work with the data using Python and ASE. Browse the databases by clicking the [browse] links below or download the databases and explore them using ASE tools:
Other links:
CAMD-Web software
All CMR databases are licensed under a
Creative Commons Attribution-ShareAlike 4.0
International License





All projects¶
- Uncertainty-aware electronic density-functional distributions
- Atoms and molecules
- Solid-state data by Tran, Stelzl and Blaha
- Example
- Chemisorption of gas atoms on one-dimensional transition-metal halides
- Impurities in 2D Materials Database
- A systematic computational database for one-dimensional materials
- Database of ABSe3 materials
- Bondmin optimization algorithm
- Definition of a scoring parameter to identify low-dimensional materials components
- Database of A2BCX4 materials
- Ground state orderings in icosahedral Ag-Au nanoparticles
- Screening for PV and PEC materials using the OQMD database
- One, two and three component references from OQMD
- Database of ABS3 materials
- Benchmark adsorption and surface energies with RPA
- Database of Organic donor-acceptor molecules
- Database of ABX2 materials
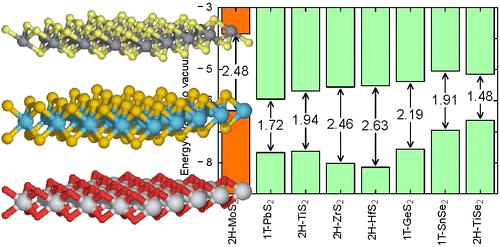
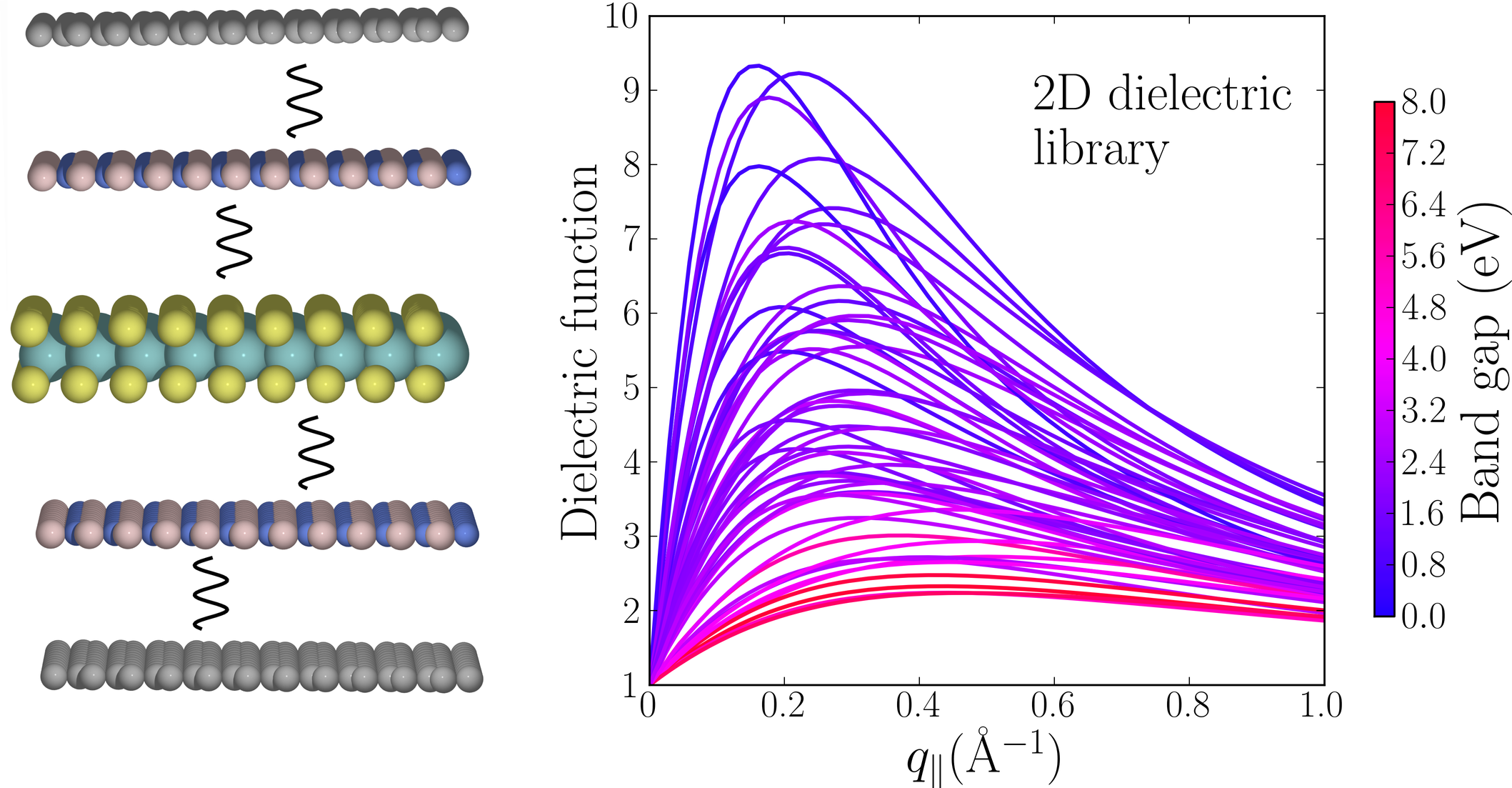
- Monolayer transition metal dichalcogenides and -oxides
- Van der Waals heterostructures
- Organometal Halide Perovskites
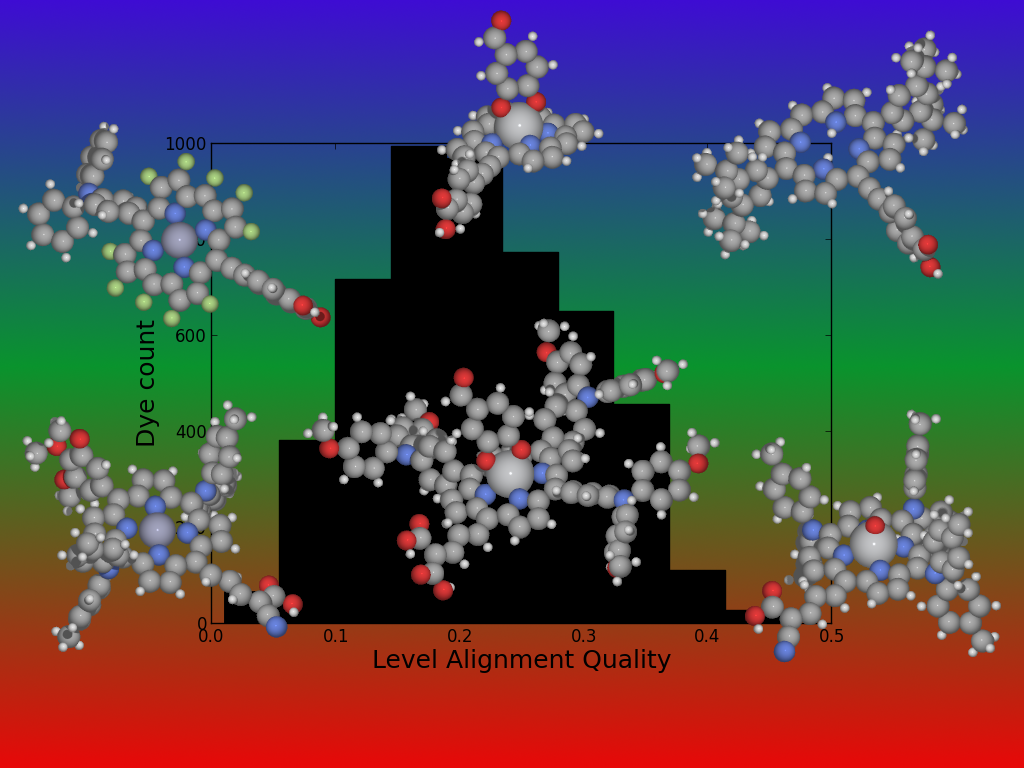
- Porphyrin based dyes
- New Light Harvesting Materials
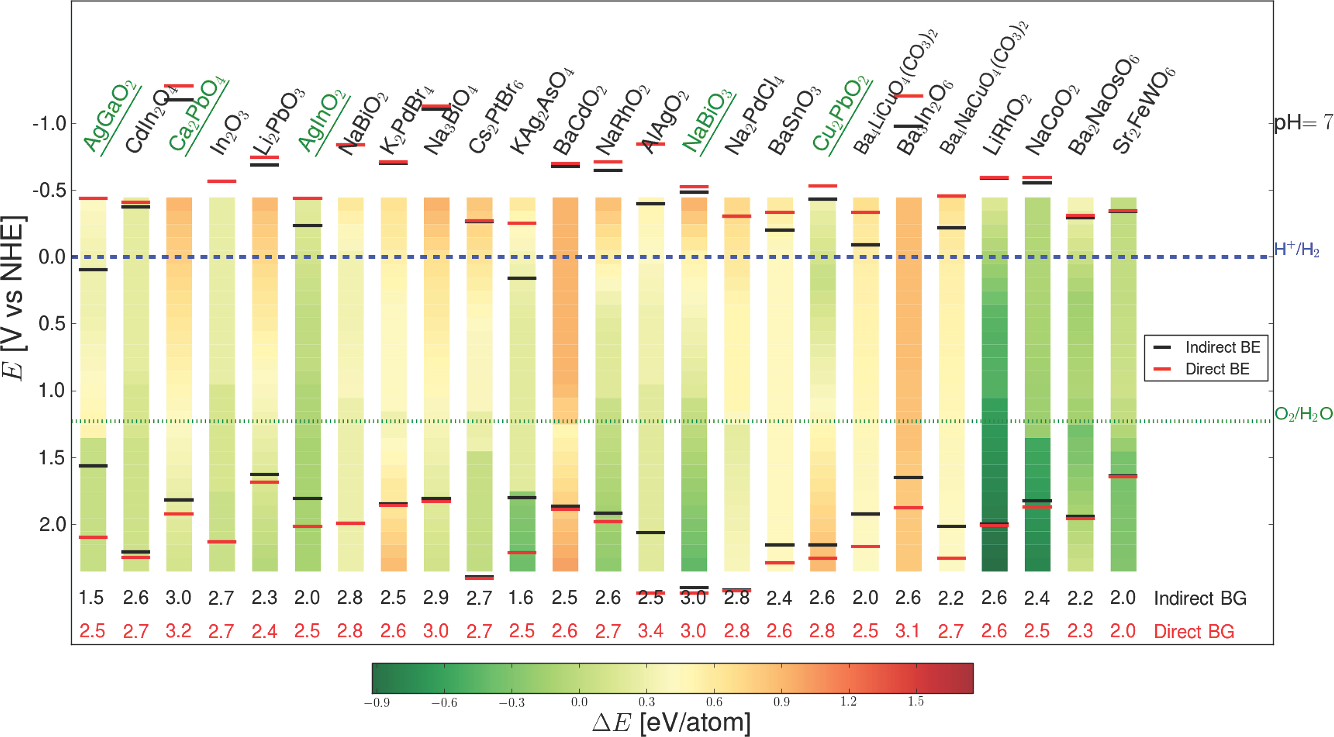
- Perovskite water-splitting
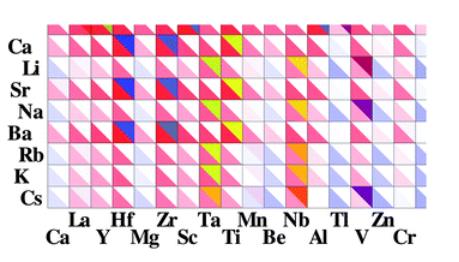
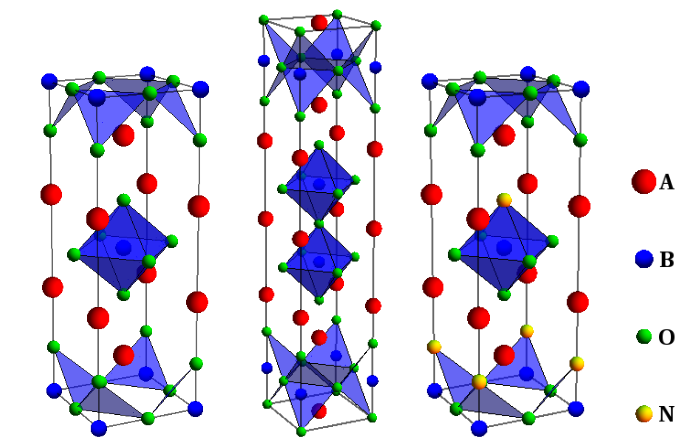
- Low symmetry perovskites
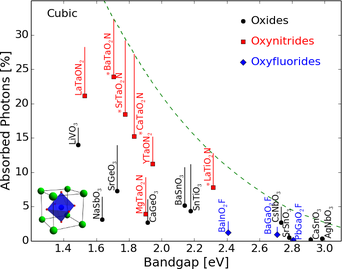
- Absorption spectra of perovskites
- Functional Perovskites
- Bayesian error estimation functionals
- CatApp database
- Data for reproducing HTGW Paper
- One and two component references from OQMD
About this web-page¶
Older databases¶
- Banchmark: DeltaCodesDFT
- Benchmark: the performance of semilocal and hybrid density functionals in 3d transition-metal chemistry
- Benchmark: adsoption energy of atomic oxygen and carbon on fcc111
- Benchmark: compression energies of bulk fcc and rocksalt
- Pseudopotentials for high-throughput DFT calculations
- G2/97